Summary
We report a childhood case of thalamic atypical extraventricular neurocytoma that progressed to highly anaplastic ganglioglioma after 8 years of dormancy after subtotal resection and chemotherapy. The neurocytoma displayed immunoreactivity only for synaptophysin, β-catenin, S100, and CD56. The ganglioglioma acquired strong immunoreactivity for chromogranin, glial fibrillary acidic protein, neuron-specific enolase, and p53 and showed a very high proliferation rate approaching 50% in some areas. Tumor transformation was associated with overexpression of components of the sonic hedgehog and Wnt developmental signaling pathways, which are known to regulate tumor-initiating cells in malignant brain neoplasms.
- Introduction
Central neurocytoma (CN) is a rare and benign intraventricular tumor [1] usually affecting young adults [2]. Shortly after delineation of this entity, similar neoplasms were described in other parts of the central nervous system and designated extraventricular neurocytomas (EVNs) [3]. Although most CNs behave as low-grade neoplasms, a significant proportion of EVNs may acquire some histologic features of atypia and display aggressive clinical behavior [4]. The atypical variant of EVN is characterized by increased mitotic activity, vascular proliferation and necrosis, and a tendency to a wider spectrum of neuroectodermal differentiation, including the relatively common development of ganglionic and astrocytic components [5]. However, these additional histologic patterns of differentiation are not associated with any change in the grade of EVNs [5]. We report pathological and molecular findings of an EVN that progressed to anaplastic ganglioglioma over the period of 8 years after subtotal resection of the primary tumor at the age of 8 months.
- Clinical history and methods
The patient was initially admitted to the hospital in early 2006, at 8 months of age, for resection of a left thalamic tumor involving the proximal midbrain and causing obstructive hydrocephalus (Fig. 1A ). She underwent a biopsy, followed by subtotal resection, intraventricular shunt placement, and chemotherapy (72 weeks of vincristine, cis-platinum, cyclophosphamide, and etoposide). Her surveillance brain magnetic resonance imaging demonstrated residual nonprogressive tumor measuring 1.8 × 2.6 × 2.5 cm in the left thalamus, with extension to mesencephalon and pons. Postoperatively, she was left with a right hemiparesis, seizures, speech problems, and visual field deficits and attended special education classes until fourth grade.
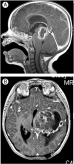
Fig. 1
Gadolinium-enhanced magnetic resonance imaging. A, Large, relatively well-demarcated, ring-enhancing thalamic tumor at the age of 9 months. B, Recurrent tumor involving the wall of postsurgical cavity and replacing the entire thalamus.
Approximately 9 years later, she was admitted to the hospital after several weeks of worsening right-hand weakness associated with a 1-week history of headache. Computed tomography showed expansion of the tumor to most of the basal ganglia (to approximately 5.6 × 4.4 × 5.3 cm), surrounding edema with left-to-right midline shift, and obstructive hydrocephalus (Fig. 1B). She underwent 2 additional craniotomies for extensive subtotal removal of the tumor with the residual lesion remaining in the left basal ganglia and brainstem. She started 33 fractions of focal radiation to the tumor area (5940 cGy) and daily oral temozolomide (90 mg/m2). She died of relentless progression of the tumor, approximately 8 months after the last surgery and 3 months after completing radiotherapy. There is no known relevant family history of brain tumors, childhood cancer, genetic syndromes, or neurodevelopmental delay. BRAF V600E, mismatch repair gene mutation, and histone H3.3 mutation testing results were negative.
The tumor tissue from each resection was routinely processed for paraffin embedding and stained with hematoxylin and eosin. Selected sections were immunostained for some or all of the listed tissue antigens with commercially available primary antibodies (Table), following the producer’s recommended dilutions and tissue preparations as previously reported [6]. For molecular studies, nanoString Technologies, Seattle, USA gene expression profiling methods were used as previously reported [7].
- Results
Microscopic examination of the surgical specimen from the first resection at the age of 8 months revealed a neoplasm composed of uniformly small cells with rounded nuclei that had occasional delicate nucleoli, and small rim of cytoplasm (Fig. 2A ). There was no nuclear atypia or pleomorphism. Several mitotic figures and scattered apoptotic nuclei were present in the entire specimen. The capillaries showed prominent endothelium, but no signs of vascular proliferation. Large areas of necrosis were associated with scattered, thrombosed vessels of various sizes. Neoplastic cells were positive for SYN (Fig. 2B), cytoplasmic B-Cat, S100, and CD56 (Table). The tumor was entirely negative for GFAP, NF, EMA, β-tubulin, HMB45, CHGR, and p53. MIB-1 was positive in approximately 5% of nuclei. Electron microscopy revealed tightly packed, small cells with no signs of intercellular junctions of any type or of synapse formations. The cytoplasm contained occasional Golgi apparatus, small profiles of endoplasmic reticulum, ribosomes, and rare electron-dense neurosecretory granules. Small parallel collections of microtubules were present in short neuritic processes.

Fig. 2
A and B, Specimen from the first resection. Uniformly well-differentiated small cells (A) displaying strong reaction for synaptophysin (B). C-F, Recurrence 9 years later. C, Highly anaplastic tumor displaying marked nuclear hyperchromasia, multinucleation, and prominent vascularization. D, Strong reaction for synaptophysin in binucleated ganglionic cell (arrow) and small neurocytic cells. E, Intense GFAP reaction in astrocytic component and negative ganglionic cell and neurocytic cells (arrows). F, Immunoreactivity for P53 in many nuclei.
View Large Image | View Hi-Res Image | Download PowerPoint Slide
The recurrent tumor resected at the age of 9 months revealed markedly increased cellularity, nuclear hyperchromasia, and the presence of scattered large, highly atypical multinucleated cells (Fig. 2C). There was marked vascular proliferation and extensive areas of necrosis. Scattered mitoses, up to 5 per high-power field, were present in a few small foci. The immunophenotype of the neoplasm changed dramatically (Table), and in addition to widespread immunoreactivity for S100, SYN (Fig. 2D), B-Cat, and CD56, many atypical cells expressed GFAP (Fig. 2E) and CHRG, indicating the emergence of new, malignant astrocytic and ganglionic components. p53 was positive in the nuclei of all the neoplastic cells (Fig. 2F), and MIB-1 increased up to 20% to 50% of the cell population. The recurrent tumor still contained foci of neurocytoma similar to that in the first resection. However, the cells were slightly larger, and showed increased nuclear atypia and frequent mitoses. Transitional cell types between neurocytes and large highly atypical ganglionic cells were commonly seen.
A NanoString gene expression custom codeset identified genes preferentially enriched in primary or recurrent tumor samples (Fig. 3). GFAP and TP53 were highly expressed in both primary and recurrent lesions. Of particular interest, TWIST1 and EOMES, both representing components of the epithelial-mesenchymal transition, were highly expressed in the primary sample, whereas sonic hedgehog (Shh) and Wnt signaling pathways (GLI3, CTNNB1) were enriched in the recurrent sample.
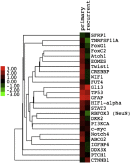
Fig. 3
Gene NanoString gene expression custom codeset, showing differentially enriched components of the Shh (GLI3) and Wnt (CTNNB1) signaling pathways in the recurrent anaplastic ganglioglioma, and enrichment of epithelial-mesenchymal regulators (TWIST1, EOMES) along with STAT3 in the primary CN. Expression profiles are displayed as fold-change ranging from a 3-fold reduction (bright green) to a 3-fold increase (bright red).
- Discussion
CN and EVN are extremely rare in the pediatric age group and are thought to represent only 0.2% of all pediatric brain tumors [8]. Large multicenter studies have determined the mean age of onset of both CN and EVN as approximately 30 years [2]. EVNs most commonly develop in the frontal lobe (34%) [9]. Thalamic EVNs have been described only twice [9]. CNs are usually World Health Organization grade II tumors [10]; however, EVNs more frequently contain atypical histologic features such as necrosis, vascular proliferation, increased mitotic activity, and MIB-1 in more than 2% of cells [[4], [5]], all associated with more adverse clinical outcomes [11]. EVNs also more commonly display glial markers such as GFAP and ganglionic patterns of differentiation. von Deimling et al [12] demonstrated the expression of glial and neuronal markers in the same CN cell, indicating that these cells retain the ability for multipotential differentiation, and proposed that CN may originate from a subependymal progenitor cell. The in vitro work by Sim et al [13] has determined that CN cells remain environmentally biased to neuronal lineage. However, CN cells can undergo phenotypic change and glial differentiation when removed from the subependymal environment, supporting the hypothesis of von Deimling et al [12]. Although EVNs are more likely than CNs to contain a gangliocytic component, transformation of EVN to anaplastic ganglioglioma has never been described.
The recurrent tumor displayed markedly increased cellular atypia accompanied by greatly increased proliferation rate and p53 expression in all cells (Table). The presence of transitional cells between neurocytes and ganglionic cells and the emergence of a malignant glial phenotype resulted in a mixed tumor with features of anaplastic ganglioneurocytoma and malignant astrocytoma, justifying the designation of anaplastic ganglioglioma. This case is a unique example of parallel malignant transformation of both neuronal and glial components, a phenomenon exceedingly uncommon in nonradiated gangliogliomas [14].
Malignant progression in many tumors is often associated with mutations in cell-cycle regulatory genes such as TP53 [15], also strikingly overexpressed in the malignant neurocytic, astrocytic as well as ganglionic phenotypes of the recurrent tumor in our patient. p53 is consistently negative in CNs and EVNs as well as in the low-grade ganglionic tumors. However, Pandita et al [15] demonstrated the development of p53 immunoreactivity selectively in the malignant portions of gangliogliomas. Although p53 NanoString gene expression profiling did not correlate with our immunohistochemical analyses, these differences may be reconciled by the greater level of sensitivity that is not available with immunohistochemistry. Of particular interest in this patient is enrichment of developmental signaling pathways that have previously been shown to regulate tumor-initiating cells in malignant brain tumors [16]. The activation of key components in Shh and Wnt signaling pathways (GLI3, CTNNB1) may contribute to driving treatment-refractory tumor-initiating cells. CTNNB1 codes for β-catenin, the key downstream effector protein in the Wnt pathway. Gli3 functions as a transcription factor in the Shh pathway. Previous transcriptome data from CN [17] suggest that similar oncogenic pathways may operate in EVN. The presence of key components of the epithelial-to-mesenchymal transition process (TWIST1, EOMES) in the primary sample indicated the potential for malignant transformation in the primary tumor. Both TWIST1 and EOMES have been shown to promote an aggressive and metastatic phenotype in primary cancers [18]. Consequently, TWIST1 and EOMES may potentially serve as drivers of malignant progression in our primary tumor.
In summary, we describe a unique pediatric case in which an atypical residual EVN displayed accelerated growth many years after partial resection, due to the development of a dominant component of anaplastic ganglioglioma. Although CNs are benign tumors, EVNs differ due to increased frequency of histologic atypia and potentially worse prognosis after partial resection. Our molecular data suggest potential biomarkers that may predict malignant transformation of EVN.